1. Introduction
Reducing anthropogenic greenhouse gases such as carbon dioxide from the atmosphere is a critical issue throughout the entire world. During the last few decades, CO2 emissions have grown extensively as a result of developing technologies and economies that utilize fossil fuels.1) Given that CO2 is believed to be a primary contributor to climate change, its concentration in the atmospheric air should be decreased. Several carbon capture and sequestration techniques have been proposed to tackle this issue, which eventually requires CO2 conversion to other forms of energy or chemicals.2-5) In recent years, intensive research has been conducted on CO2 conversion technologies that directly transform CO2 into useful energy or value-added products, among which high-temperature co-electrolysis has shown substantial potential.6-8)
In our previous study, we used solid oxide cells to perform high-temperature co-electrolysis of CO2/steam mixtures in order to produce syngas.9-11) The presence of CO2 and H2O makes the fuel-electrode reacting environment complicated in a way in which electrochemical (i.e., electrolysis) and thermochemical (i.e., reverse water gas shift (RWGS)) reactions proceed simultaneously. It has been found that steam is a primary reactant in electrolysis, while carbon dioxide is reduced predominantly by the RWGS. To enhance CO2 conversion, some researchers suggested that fuel-electrode materials need to be modified so that the activation energy for CO2 reduction decreases. Among various methods, infiltrating noble metal catalysts into fuel-electrode supported cells is a promising technique to provide sufficient performance by enhancing CO2 conversion and the ability to control syngas production. A number of precious and transition metals, such as Pt, Ru, Rh, Pd and Fe, have been proposed as catalysts for RWGS, among which Pd and Fe have been found to be catalytically effective.12,13)
In this study, a metallic catalyst, palladium or iron, to promote RWGS, was employed in a fuel-electrode with 5 × 5 cm2 solid oxide cells using a wet infiltration technique. The long-term stability of the catalyst-infiltrated fuel-electrode supported solid oxide cells was examined at 800°C during 350 h of co-electrolysis operations. Both the electrochemical and gas-production performances were monitored in the galvanostatic operation at 0.3 A/cm2. The stabilities of Pd-and Fe-infiltrated cells were compared, and the sources for degradation were examined.
2. Experimental Procedure
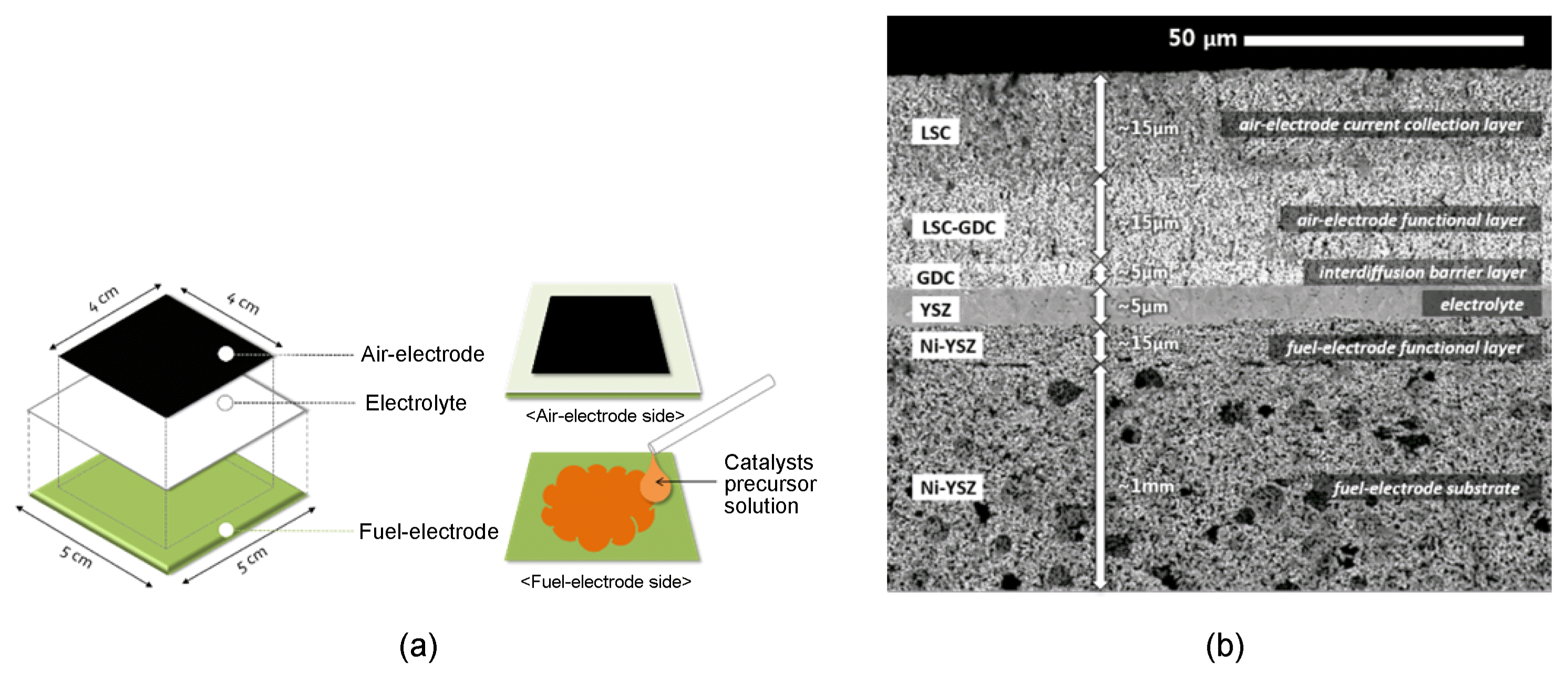
The planar and fuel-electrode supported cells used in this study were comprised of a Ni-YSZ substrate, Ni-YSZ fuel-electrode functional layers, a YSZ electrolyte, a GDC inter-diffusion barrier layer, La0.6Sr0.4CoO3-δ (LSC)-GDC air-electrode functional layers and LSC air-electrode current collection layers. The fuel-electrode substrate was prepared by tape-casting and successive lamination. For manufacturing the slurry for tape-casting, NiO (Sumitomo Metal Mining, Japan), YSZ (8 mol% Y2O3 stabilized ZrO2 (TZ-8Y), Tosoh Corp., Japan) and a poly(methyl methacrylate) (PMMA) pore-forming agent were mixed with a dispersant (HypermerTM KD-6, Croda, United Kingdom), binder (ethyl cellulose, Sigma-Aldrich, USA) and plasticizer (dibutyl phthalate, Junsei chemical, Japan) in ethanol and toluene, and ball-milled for 24 h. Tape-casted green-tapes were subsequently compacted by uni-axial pressing at 10 MPa and 75°C, producing the ~ 1.3 mm thick fuel-electrode substrate. The pastes for the fuel-electrode functional layers, electrolyte, interdiffusion barrier layer, air-electrode functional layers, and air-electrode current collecting layers were prepared by mixing corresponding powders with the dispersant, binder and plasticizer in solvent (α-terpineol, Kanto chemical, Japan) and using a planetary mill. The fuel-electrode functional layers and electrolyte were screen-printed on the substrate and co-sintered at 1300°C. The interdiffusion barrier layer was screen-printed on top of the YSZ electrolyte and sintered at 1250°C. Subsequently, the air-electrode functional layers and air-electrode current collecting layers were screen-printed and sintered at 950°C. The size of the unit cells was 5 cm × 5 cm, and the effective electrode area was 4 cm × 4 cm.
Catalyst nanoparticles were infiltrated to the porous fuel-electrode substrate using their precursor solution mixed with urea. To remove trapped air inside the fabricated unit cells, they were thermally treated at 400°C for an hour prior to infiltration of the catalyst nanoparticles. The precursor solutions were mixed with urea (Sigma-Aldrich, USA), which resulted in a molar ratio of urea to cations of 10. Infiltration was performed by applying the formulated solution to the fuel-electrode substrate surface, as shown in Fig. 1(a), which was facilitated by vacuum treatment. The infiltrated cells were thermally treated at 80°C for 2 h to decompose the urea, followed by secondary thermal treatment at 400°C for an hour to remove organic compounds. The infiltration and thermal treatment steps were repeated to obtain a designated amount of catalyst loading prior to in-situ crystallization at 800°C during cell operation. Fig. 1(b) shows the prepared fuel-electrode supported cells impregnated with the catalysts. Further details of cell fabrication and wet infiltration procedure were discussed in our previous study.10,11)
The long-term stability for co-electrolysis operations was characterized by placing the prepared cells between metallic jigs and forming hermetic gas-sealing conditions with the aid of glass-ceramic sealants. Pt mesh and Ni foam were used for current collection for the air-electrode and fuel-electrode, respectively. The cell-sealant-jig assemblies were installed in a hot furnace at 850°C with compression of 30 kgf for 5 h to obtain hermetic sealing. Then, the NiO fuel-electrode was reduced to Ni by feeding of wet hydrogen, which simultaneously resulted in M(Pd or Fe)-Ni bimetallic alloys. The long-term co-electrolysis operations during 350 h were performed at 800°C using the feed gas comprised of 10% H2O, 50% CO2, 10% H2. The flow rates of both the air and feed gas were maintained constant at 200 sccm. The electrochemical impedance spectra (EIS) were obtained at every 100 h using a frequency response analyzer and potentiostat (Solartron 1260/1287, Solartron Analytical, United Kingdom). The gas-production was monitored by gas chromatography (Agilent 7890B, USA) connected to the tail-end of the cell-testing system. Further details of the performance characterization procedure were discussed in our previous study.9)
3. Results and Discussion
The high-temperature co-electrolysis process has various economic, durability, and reliability problems due to its high operating temperatures. Particularly, deterioration problems of membranes, electrodes and stacks of the co-electrolysis system are much more serious than those of SOFCs operating in similar environments. Hence, design optimization of materials and structures for co-electrolysis stacks that can cope with these problems is needed. Previous studies on the long-term stability of high-temperature electrolysis cells have been reported mainly for the electrolysis of water vapor,7,14,15) but rarely for the CO2 electrolysis.7) Major deterioration phenomena reported so far are performance degradation of the fuel electrode due to water vapor,16,17) agglomeration of the small catalyst particles and carbon deposition on the catalyst surface.18,19) Reduced active sites for the electrochemical/thermochemical reaction by impurity contamination from reactant gas,20) and delamination at the interface between the air electrode and the electrolyte 6,21) have been also reported as deterioration origins. As listed above, most studies on the long-term stability of high-temperature co-electrolysis cells have been generally carried out via electrochemical AC impedance analysis on the degradation of cell performance. However, given that the ultimate goal of high temperature co-electrolysis is syngas production, monitoring product gas composition during the long-term operation of the co-electrolysis system is necessary in addition to the electrochemical characterization. Furthermore, in the case of the metal catalyst incorporated fuel electrode, its catalytic activity for the CO2 conversion is easily degraded by the agglomeration of the nano-sized metal catalyst, and thus the long-term stability of catalyst-infiltrated fuel electrode should be scrutinized. Hence, in this study, we conducted a characterization of the electrochemical performance and syngas production yield of co-electrolysis cells and evaluated the long-term stability of the catalyst-infiltrated fuel electrode with respect to the effective reaction sites altered by the coarsening of the nano-catalyst and impurity deposition under high temperature co-electrolysis conditions.
Figure 2 shows the temporal variation of a unit cell voltage measured for 350 h at a constant current of 0.3 A/cm2. As shown in Fig. 2, the cell voltage of both Pd- and Fe-infiltrated solid oxide cells continuously increases throughout the measurement time. Since the overvoltage is directly related to the amount of electric energy supplied from external sources, the gradual increase of the overvoltage leads to a loss of the co-electrolysis efficiency. Therefore, it is very important to suppress the gradual increase of the overvoltage that can occur under the severe operating conditions of high temperature co-electrolysis. According to our test results in Fig. 2, the electrochemical degradation rate after 350 h was about 0.93% for the Pd-infiltrated cell and about 2.42% for the Fe-infiltrated cell, all of which showed a relatively low degradation rate, indicating the possibility of stable operation. On the other hand, the relatively higher degradation rate of the Fe-infiltrated cell can be attributed to the lower tolerance of Fe for carbon coking compared with Pd. Pd is known to have a higher coking resistance to carbon than other metal catalysts and is already used as a general co-catalyst for conventional Ni-based fuel electrodes.22,23) Hence, we can conclude that under the same operating conditions, the Pd-infiltrated cell has a higher resistance to carbon deposition than the Fe-infiltrated cell, thereby showing a relatively low electrochemical degradation rate. These results are consistent with the changes in syngas production yield that will be discussed later.
Figure 3 shows the electrochemical impedance spectra (EIS) obtained in OCV conditions at every 100 h during 350 h of co-electrolysis operations. As shown in Fig. 3, the ohmic and polarization resistances of both the Pd- and Fe-infiltrated cells slightly increased, but the change was negligible, as was previously confirmed in Fig. 2; the overvoltage increase rate was less than 1% per 100 h. Interestingly, both cells exhibited high polarization resistance near the 10−1 to 100 Hz range, which is generally assigned to the electrochemical reaction limited by the fuel electrode reaction and/or the mass transfer reaction within the electrode structure. 9)
Figure 4 shows the temporal variation of produced gas composition that was measured for 350 h under galvanostatic co-electrolysis operations at 0.3 A/cm2. According to the results, syngas-production stabilities of both the Pd- and Fe-infiltrated cells did not deteriorate significantly during the co-electrolysis operation, but the CO concentration gradually decreased, indicating a decrease in the CO2 conversion rate, although it was not significant. This gradual decrease in the CO2 conversion rate was more prominent in the Fe-infiltrated cell, which can be attributed to the fact that Fe is less resistant to carbon coking than Pd. This higher carbon coking tolerance of Pd allows long-term reliability of electrode activity in hydrocarbon environments by suppressing deposition of carbon produced from the general Boudouard reaction (disproportionation reaction of carbon monoxide into carbon dioxide and graphite). Due to the difference in the carbon deposition resistance between Pd and Fe, the deterioration rate of synthesis gas production characteristics was about 0.81% for the Pd-infiltrated cells and 4.82% for the Fe-infiltrated cell. Hence, we concluded that the incorporation of Pd into the fuel electrode is an effective way to secure the long-term stability of a high temperature co-electrolysis system.
4. Conclusions
We analyzed the long-term stability of high temperature co-electrolysis cells for electrochemical/thermochemical syngas production: 5 cm × 5 cm fuel electrode supported cells were fabricated via conventional multilayer ceramic processing, and additional Pd- or Fe-based nano-catalysts were infiltrated into the Ni-based fuel electrode to improve CO2 conversion. The overvoltage of the co-electrolysis cells and the produced gas composition were analyzed for 350 h under a constant current at 0.3 A/cm2. Both Pd- and Fe-infiltrated cells showed a low electrochemical degradation rate of less than 1% per 100 h. The Pd-infiltrated cells had a degradation rate of 0.81% during 350 h, while the Fe-infiltrated cells had 4.82%. The lower degradation rate of the Pd-infiltrated cell can be explained by the relatively higher carbon coking tolerance of Pd catalysts.