1. Introduction
In the production of artificial bone materials that can substitute autogeneous bone, we attempted to develop calcium phosphate compound (CPC) bone cement1-5) in order to improve the material’s toughness instead of using calcium phosphates for the source of hydroxyapatite (HAp, Ca5(PO4)3(OH)).6,7) Since the first CPC,1,2) consisting of an equimolar mixture of tetra-calcium phosphate and dicalcium phosphate anhydrous, was developed in 1986, many kinds of CPC powders have been developed, mainly by using TCP and/or TTCP.8-15) β-TCP is the low-temperature phase in the CaO-P2O5 phase diagram and is known to be resorbable in vivo with new bone growth, replacing the implanted β-TCP. Tricalcium phosphate (TCP) is one of the most important biomaterials based on phosphates, and it is currently recognized as an artificial bone material that significantly simulates the mineralogical structure of bone. Theoretically, resorbable TCP is an ideal implant material. After implantation, TCP degrades with time and is replaced with natural tissues. It leads to the regeneration of tissues instead of their replacement, and thus solves the problem of interfacial stability. According to Metsger et al.,9) tribasic calcium phosphate is a nonstoichiometric compound often bearing the formula of hydroxyapatite [Ca10(PO4)6(OH)2]. TCP is a resorbable temporary bone space filler material. 12,13)
In the previous research,3-5) a nano-crystalline calcium phosphate (CaP) was prepared at 37°C in an aqueous solution of active Ca(OH)2 and H3PO4. We reported the crystal growth of CaP slurries and the mixing effect of the crystallites to increase the toughness of the artificial bone.5) In this research, we focused on the cement hardening reaction in CaP bone cement formulation using aqueous poly-phosphoric acid (PPA) solution, Ca(H2PO4)2·H2O (MCPM), and CaSO4·1/2H2O (CSH). We used a micro-crystalline CaP paste, which was prepared at 37°C in an aqueous solution of active Ca(OH)2 and H3PO4. The crystal growth was carried out at 37°C for 96 h. The bone cement blocks were prepared by setting the temperature and time of the process. Mechanical strength with microstructural variation was measured for the bone cement blocks. The used precursor powders were evaluated by using field emission scanning electron microscopy (FESEM) and X-ray diffraction (XRD) as reference materials for the phase analysis during the cement hydrated hardening reaction.
2. Experimental Procedure
2.1. Precipitation of calcium phosphate phase using Ca(OH)2 and H3PO4 in water
The CaP precipitates were prepared at pH 5.0 in a CO2-free air atmosphere by a conventional wet method3-5) using Ca(OH)2 and H3PO4. As precursors for Ca2+ and H3-xPO4 ions, we used highly active Ca(OH)2 and H3PO4 (AP grade, Aldrich, USA) in DI water. The highly active Ca(OH)2 was obtained through the hydration of CaO, in which CaO was prepared by the calcination of CaCO3 [(Alkaline analysis grade, Aldrich, USA)] at 1150°C for 4 h. The Ca(OH)2 powders were stirred in 1000 cc of DI water to prepare the ionized aqueous solution of Ca2+. The obtained ionized Ca2+ solution was used as a base for the titration process. The CaP slurry was prepared using the simultaneous titration method with peristaltic pumps (Masterflex, Cole-Parmer, USA), a water bath (Boekel, USA), and a pH controller (Bukert 8280H, Germany). The temperature of the main reactor was digitally controlled to within 0.1°C. During the precipitation, the reaction solution in vessel was maintained at 37°C and at pH 5.0 ± 0.1 under stirring. The CaP slurry pastes were named as mDCPD-0, indicating microcrystallites of the DCPD phase from XRD analysis. For the crystal growth, the mDCPD-0 slurry solution in a beaker sealed with vinyl film was kept in a drier at 37°C for 96 h, and the obtained CaP solution was named as mDCPD-96. It was filtered and dried at 60°C for bone cement formulation.
2.2. Fabrication of DCPD bone cement blocks
The total amount of the above mixture was controlled as 2.6676 g. Aqueous poly-phosphoric acid solution was prepared by stirring 1.6667 g of DI H2O with 0.000394 g of poly-phosphoric acid [PPA, 115%, Sigma-Aldrich, Regent Grade]. As one of notations, P1 indicates a typical amount of PPA, 0.000394 g of poly-phosphoric acid with 1.6667 g of DI H2O. P2 and P0.8 indicate that the amount of PAA was two times [0.000788 g] and 0.8 times [0.000315 g] that of the P1 amount, respectively. The hardening time was controlled from 0 min to 7 min and noted as t0 to t7, respectively.
The mixture powders of mDCPD, CSH, and MCPM were placed into a dental rubber bowl and aqueous PAA solution was dropped into the mixtures by using a pipet under mixing with a plastic dental spatula. This process was done quickly within a minute, and then, the mixture was put into a 1.4 cm Φ stainless steel mold using a spatula within a minute. Then, a 1 kg steel drum (weight) was loaded on the upper punch of the mold for a minute, and then, the mold was hydrostatically pressed at 4 ton for 20 s. The sample size of mDCPD-based bone cement block was 1.4 cm Φ × 8 mm t after hardening process at 37°C for 5 min, and then, the sample block was taken out for the UTM measurement. The calculated net hydrostatic pressure was 2.6 ton/cm2. In these DCPD-based cement blocks, we did not apply polyethylene glycol bubbles as porogen to build up pores in the bone blocks.
2.3. Characterization
The crystal phase for the crushed powders was confirmed using XRD (Bruker, M18XCE). The XRD pattern was measured for the prepared mDCPD dry powders, Ca(H2PO4)2·H2O (Sigma-Aldrich, MCPM), and CaSO4·1/2H2O (Sigma-Aldrich, CSH) powders. Commercial powders (Sigma-Aldrich) of β-TCP, DCPD, and CaSO4·2H2O (CSD) were used as XRD references. FE-SEM (Hitachi, S-4800) morphology was observed for the mDCPD powders and the bone cement blocks. The compressive strength for the mDCPD-based bone cement blocks was measured using a universal testing machine (UTM, DaeSan Eng, Korea). By evaluating the strength of the bone cement blocks, we investigated the effects of mDCPD powders for the crystal phase transformation during bone cement reaction.
3. Results and Discussion
3.1. XRD for mDCPD powders and bone cement blocks
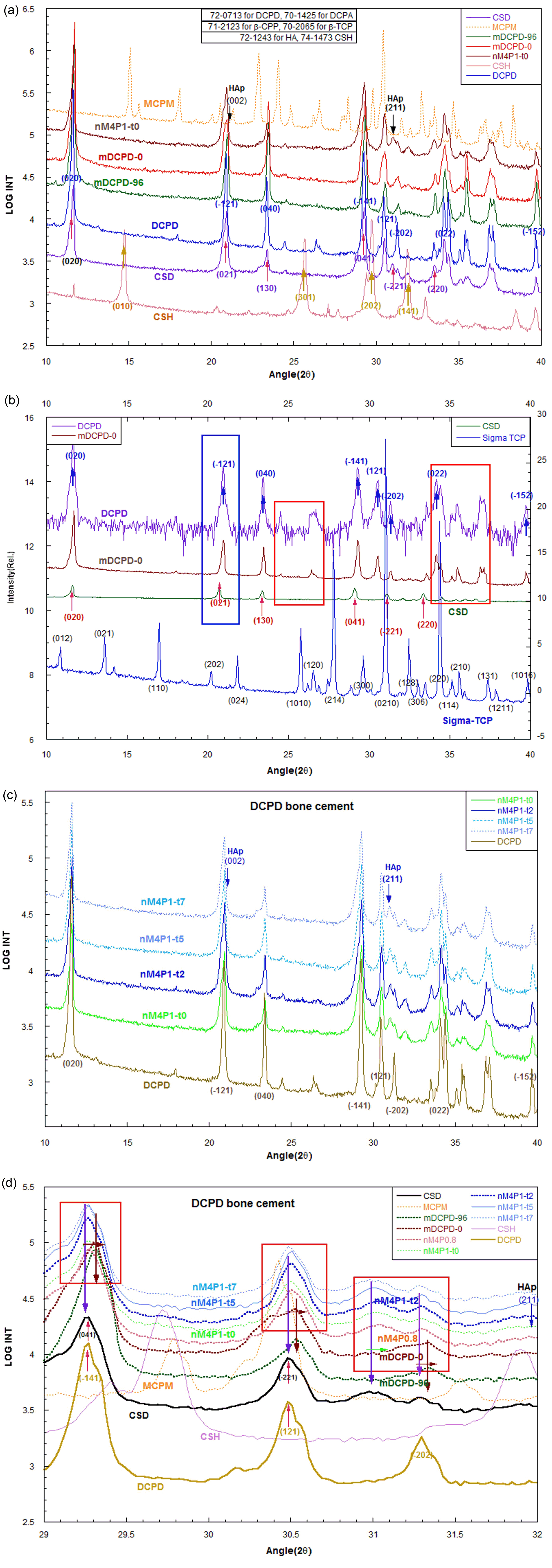
To increase the mechanical strength of the bone cement block, we used the DCPD powders of mDCPD-0 and mDCPD-96, in which the powder samples were prepared at 37°C and kept at 37°C for 96 h, respectively. It is noted that the amount of [Ca2+] in these samples corresponds to higher concentration above the saturation line in the Ca-pH phase diagram,1,2) indicating a log [Ca2+] value of −1.84.
As shown in Fig. 1(a), the main phases of mDCPD-0 and mDCPD-96 are identified as DCPD or apatitic TCP19-22) (Ca9(HPO4)(PO4)5OH) through XRD analysis. The XRD pattern for mDCPD [mDCPD-0 and mDCPD-96] is close to that of brushite,18) corresponding to that of commercial DCPD powders (Sigma-Aldrich). In Fig. 1(a), (b), the crystal phases of mDCPD-0 and mDCPD-96 are very close to those of DCPD in the entire range. The blue rectangle in Fig. 1(b) shows that the (−121) peak in DCPD is almost same as that of mDCPD-0. The (021) peak of CaSO4·2H2O is located at a lower angle compared to the similar peak of mDCPD-0, but above 2θ = 25°, the spectra shows highly different XRD patterns. Two red rectangles show similar XRD spectra for DCPD and mDCPD-0, but at higher angles above 2θ = 25°, CaSO4·2H2O shows the different XRD spectra.
The overall XRD spectra patterns in mDCPD-0 and mDCPD-96 in Fig. 1(a) are close to that of DCPD. From these results, the CaSO4·0.5H2O powders in the bone cement formulation may just contribute to the cement powder interaction by mDCPD and DCPD on the surface of the mDCPD powders. In the DCPD cement reaction, the hydration effect of CaSO4·0.5H2O powders will be discussed later by using XRD, FESEM, and mechanical strength analyses. Fig. 1(c), (d) show the bone cement blocks, in which mDCPD-0 powders blended with MCPM, CSH, and PPA solution were hydrostatically molded under 4 tons. The samples were hardened from 0 min to 7 min, and the samples were named from nM4P1-t0 to nM4P1-t7, respectively. With the increase in the hardening time, we could confirm the existence of (211) peaks corresponding to HAp. If we put the bone block samples into a simulated body fluid (SBF) solution, the HAp phase will be shown clearly with the increase in the holding time, as has been reported previously.23)
Figure 1(d) shows fine XRD spectra between 29° and 32°, and from the rectangle regions, we can confirm the small lattice variation in nM4P1-t cement blocks with increase in the hardening time. The lattice spectra of the cement blocks are at a similar position to those of DCPD, but with increase in hardening time, we can observe the weak appearance of the (211) HAp phase. It is noted that the spectra pattern is very similar with that of CSD, which can be transformed from CSH. CSH may play an active role in the morphological formation of bone cement, which is comprised of MCPM, mDCPD, CSH, and PAA solution.
The CaP powders prepared at 37°C could be nano-crystalline or micro-crystalline according to the concentration of Ca2+ and/or PO43− and pH values.3-5) Previously, we reported the preparation4) of nano-crystalline CaP powders and the crystal growth with the temperature and holding time. In this research, the FE-SEM results in Fig. 2 show that the mDCPD powders were microcrystalline, not nano-crystalline.
3.2. FE-SEM
Figure 2 shows the FE-SEM microstructures for the mDCPD-0, mDCPD-96, MCPM, and CSH samples. In Fig. 2(a), mDCPD-96 shows gradual crystal growth of hexagonally lamellar crystallites during 96 h at 37°C. From the microstructural view, the crystallites in mDCPD-96 were grown ~ 20-30% from mDCPD-0. Fig. 2(b) shows the nano-crystalline powders4) formed at 37°C. Full amount of nano-crystalline powders were observed in a homogeneous precipitation at 37°C. In some experimental batch, nano-crystalline powders were occasionally observed with microcrystalline DCPD powders, as shown in the right image of Fig. 2(b). In fact, traces of the nano-crystalline powders were observed in mDCPD-0 and mDCPD-96. We could occasionally observe the nano-crystalline powder aggregates in mDCPD-96, which are shown by blue rectangles in Fig. 2(a). The microstructural control of the wet prepared DCPD powders at 37°C was highly sensitive to preparation factors, such as pH and [Ca2+]. In wet DCPD powders, the preparation details may result in the homogeneous and/or heterogeneous reaction. The precipitation of CaP crystallites at pH 5.0 are sensitively influenced by the nucleation process factors, such as [Ca2+] and temperature. The concentration of H3-xPO4 affects the precipitation reaction,6) but the effects are not severe compared to that of [Ca2+]. This means that the log [Ca2+] values along the saturation curve1,2) greatly affect the precipitation behavior of CaP slurries from nano-crystalline to macro-crystalline. Fig. 2(c), (d) show the micrographs for MCPM and CSH, respectively. The particle size of MCPM is W 50-70 μm, L 80-100 μm, and T 2-4 μm. The particle dimension of CSH is W 10-20 μm, L 30-50 μm, and T 4 μm. CSH powders are composed of irregular-shaped platelets, which were prepared by the condensation of bar-shaped particles of several hundred nanometers.
As mentioned in Section 2-2, the MCPM, CSH, and mDCPD powders were mixed by using aqueous PAA solution, and then, the mixed slurry cement was put into a steel mold to make sample blocks for measuring the mechanical strength. Fig. 3 shows the micrographs for the broken particles of bone cement blocks remaining after the strength measurements. MS in Fig. 3(a) indicates that the microcrystallites of bone cement formulation are shaped by using plastic syringe, and the formulation recipe of the MS sample was the same as that for mM2P1. In Fig. 3(b), mM2P1 indicates that the bone cement slurries with normal PPA were shaped by using a stainless mold under 200 kg hydraulic pressure. In Fig. 3(a), the bone cement particles are composed of various rectangular plates with the dimension of W 10-20 μm, L 20-40 μm, and T 200 nm. The cement hydration reaction among MCPM, CSH, and mDCPD powders was activated by adding an aqueous PPA solution. It is considered that MCPM and mDCPD powders were combined on the surface of CSH crystallites, and then, the decomposition and recrystallization by PPA solution occurred. The hydration hardening reaction may result in the bone cement composite formation, in which the mechanical strength will be greatly affected.
In Fig. 3(b), the bone cement particles of mM2P1 are composed of various rectangular platelets with the dimension of W 0.5-1 μm, L 1-3 μm, and T 100 nm. The observed particle size was decreasing, probably due to the decomposition-recrystallization among MCPM, CSH, and mDCPD powders. Compared to MS sample of Fig. 3(a), the cement hardening interaction was highly inspired by using the hydraulic mold compression instead of the syringe shaping pressure.
Figure 3(c) for mM4P1 and Fig. 3(d) for mM4P0.8 show the hardening effect with the amount of aqueous PAA solution during bone cement formation. In the blocks, nM4 means the mold shaping by using < 400 kg hydraulic pressure. In Fig. 3(d), the crystallite size for mM4P0.8 was much higher than that of mM4P1, indicating that 0.8 times of the PPA amount reduced the reaction in the decomposition-recrystallization among the powders. A higher amount of PPA accelerated the reaction speed of the decomposition-recrystallization among the powders. We tried to find an appropriate PPA amount for best compressive strength values, and this will be discussed through the mechanical strength evaluation.
3.3. Bone cement interaction
In Fig. 4, the bone cement samples were labelled from nM4P1-t0 to nM4P1-t7 with the increase of hardening time. Fig. 4(a), (b) indicate mM4P1-t2 using mDCPD-96 and nM4P1-t0 using mDCPD-0, respectively. In mM1P1 or nM4P1, the amount of PPA solution was noted as P1, indicating the mixing of 1.6667 g DI H2O with 0.000394 g PPA. In the mM1P0 sample, P0 indicates that we added only 1.6667 g DI H2O without PPA. In Section 2-2, the formulated bone cement powders were mixed with a dropping liquid of aqueous PAA solution, shaped by using a stainless mold, and kept for a scheduled amount of time for hardening. The hardened block samples were extracted from the mold, and the compressive strength was immediately measured by using a UTM machine.
In Fig. 4, the cement interaction for cement blocks was evaluated with increasing hardening time, showing the grain growth in the mDCPD-based bone cement mixture blocks, prepared by using mDCPD-0 or mDCPD-96. As explained in Fig. 3, the microcrystalline crystallites of MCPM and CSH were well attached on the surface of uniformly arranged mDCPD macro-crystallites powders. The FE-SEM microstructures in Fig. 4 show the microcrystalline grains of sized on the order of micrometers. Compared to mM4P1-t2 in Fig. 4(a), nM4P1-t0 in Fig. 4(b) shows irregular shaped grains of ~ 20-50 μm, which are composed of 100-nm hexagonal crystallites. The grains might be formed through the condensation and laminated grain growth among cement components. In Fig. 4(b) showing nM4P1-t0, we can observe the grains laminated by long crystallite rods. In Fig. 4(c) showing nM4P1-t1, the grains were more highly condensed by the grown crystallite rods, and two red rectangle regions in the right-side view show the lamination condensation of thin rods, in which the thickness is smaller than that of nM4P1-t2 in the right-side view of Fig. 4(d). The thickness was estimated as ~ 100 nm for nM4P1-t1 and ~ 200 nm for nM4P1-t2, respectively.
As mentioned above, the condensation reaction occurred by the dissolution precipitation among the bone cement powders, such as mDCPD, CSH, and MCPM. The decomposition-recrystallization reaction was activated by using the typical amount, noted as P1, of PAA solution. When we mixed the rectangular micro-crystallites with CSH and MCPM micro-crystallites powders, the mixture of parallel arranged crystallites induced the laminated grain growth during the hydration hardening at 37°C for several minutes. The hardening reaction was performed from zero min to 7 min, and Fig. 4(a)-(f) show each microstructure from 0 to 7 min of hardening.
It is noted that mM4P1-t2 sample in Fig. 4(a) used mDCPD-96 powders, and the other samples used mDCPD-0 powders. From Fig. 4(a), (b), we can observe the rectangular- shaped grains, in which lamella crystallites were well condensed through the interactive reaction among powders.
In nM4P1-t0 of Fig. 4(b), the mDCPD-0 powder-based cement block was shaped under 4 ton pressure and immediately taken out for the MTS measurement. In nM4P1-t1 [Fig. 4(c)], nM4P1-t2 [Fig. 4(d)], nM4P1-t5 [Fig. 4(e)], and nM4P1-t7 [Fig. 4(f)], the sample blocks using mDCPD-0 powders were hardened under 4 ton pressure from 1 to 7 min, respectively.
For mM4P2-t2 using twice amount of P1 shown in Fig. 5, the sample block was hardened for 2 min under 4 ton pressure. The microstructures with 1-μm scale bar show the development of large laminated crystallites, and we can observe lamella crystallites of size about 100 nm on the surface of 10-μm rectangular laminates crystallites with thickness of ~ 50 nm. We can confirm that the crystallites were developed by the active cement interaction, in which the hardening continued during the mold press shaping for 2 min. The 10-μm structure images clearly show the laminated rectangular structures of 100-nm-thick crystallites. In the 50-μm and 100-μm microstructure images, we could frequently find the round irregular shapes composed of the 100-nm lamella crystallites. It is considered that such cement structures are developed by the solid-liquid interaction among DCPD, MCPM, CSH, and PAA hardening activator.
3.4. Mechanical property of bone cement block
One of research aims was to understand the cement interaction in the DCPD bone cement blocks with mechanical strength and XRD analysis. Additionally, the cement hardening effect was investigated with PAA amounts between 0.8 and 2.0.
Figure 6 shows the compressive strength with the hardening time for the cement block samples of mM4P1, nM4P1, and nM4P0.8 shown in Fig. 1. The maximum strength values were 65.1 MPa at 1 min, 54.2 MPa at 3 min, and 52.35 MPa at 2 min for mM4P1, nM4P1, and nM4P0.8, respectively. The maximum compressive strength was observed at 1 min in the nM4P1 sample [65.1 MPa, 1 min]. However, the maximum strength of mM4P1 [54.2 MPa, 3 min] was smaller than that of nM4P1 and was shown at a longer hardening time, i.e., 3 min. The amount of PAA was the same in mM4P1 and nM4P1, but was 0.8 times in nM4P0.8, which shows the maximum strength of 52.35 MPa at 2 min compared with that [54.2 MPa, 3 min] of mM4P1. From the compressive strength and hardening time, the cement samples using mDCPD-0 powders with smaller powder size show higher strength at shorter hardening time. If nano-crystalline DCPD powders are used in cement formulation, the strength can be considerably increased. In Fig. 4, the nM4P1 samples show the microstructural variation with hardening time, and the nM4P1-t1 sample shows the coagulated growth of grains condensed through the hydration reaction by PAA solution. Further, in Fig. 3(d), the nM4P0.8 sample shows similar grain growth through the hydration and condensation with a less amount of PAA. The cement blocks shown in Fig. 3, 4, and 5 exhibit dense microstructures. Fig. 6 shows the compressive strength values for a dense body with few pores.
For human bone application, the proper porosity for β-TCP bone fillers16,17,24,25) is in the range of 100-400 μm, which would indicate continuous pore channels. It is known that the porosity is roughly 75%, and the compressive strength is 2-3 MPa, which may be good for surgical applications. In mM4P1 and mM4P0.8 shown in Fig. 3(c), (d), we can observe several tens of pores, in which the pore size is under 3 μm. In mM4P1-t2 and nM4P1-t1 shown in Fig. 4(a), (c), we can find only several pores with similar pore sizes, and nM4P2-t2 in Fig. 5 shows only several larger pores from 5 to 10 μm. The compressive strength for mDCPD cement blocks in Fig. 6 was in the range of ~ 25-65 MPa during several minutes of hardening. Compared to the strength of 2-3 MPa of the β-TCP bone fillers for human bone application, the higher strength in these samples might be influenced by the cement hardening condensation in lower porosity range under ~ 10% from the SEM results. In fact, the formation of pore channels several hundred micrometers in size is a crucial factor for bone application, and we will report the porosity effect on the compressive strength through further research.
In CaP cement, the (211) phase indicates the existence of the HA phase,16) which can be precipitated in this cement reaction.
The inner graph in Fig. 6 shows the XRD intensity for the (211) phase, which clearly appeared with the increase in hardening time until 7 minutes. In spite of the error width in XRD intensity, it is believed that the HA phase was gradually developed with hardening time during the cement interaction among CSH, mDCPD, and MCPM with the addition of APP solution. From Fig. 1, the hardened cement shows the formation of the DCPD phase, not CSD. CSH powders are well dissolved23-25) during hardening by using aqueous PAA, compared to mDCPD. Therefore, there will be a higher concentration of [Ca2+] and [PO43−] in the CaP-based formulation, and finally, HAp phase will be formed with an aqueous reaction such as SBF solution.
4. Conclusions
The micro-crystalline DCPD powders prepared at pH 5.0 and 37°C were kept at 37°C for 96 h. The crystal growth and crystallite growth in the matrix of the DCPD powders greatly affected the cement interaction with MCPM, CSH, and aqueous solution of PAA. We could confirm the dissolution and recrystallization on the surface of micro-crystalline DCPD powder crystallites. The cement interaction was much higher in the as-prepared DCPD powders, compared to the DCPD powders grown for 96 h, and thus contributed to the increase in the mechanical strength in the DCPD bone cement.