1. Introduction
Silica is a chemical ionic conjugate (SiO2) of silicon (Si) and oxygen (O) and is the main component of rock, earth and sand; it is the main constituent of the earth’s crust, accounting for more than 28% of the crust. Silica particles, due to their material characteristics of having rich resources, excellent chemical stability, high strength and good adsorption characteristics, have been used in a wide variety of applications, including as heat insulators, noise suppressors, eczema agents, adhesives, inks and paints, aerogels, coatings and fillers for tires subjected to severe wear.1-2) Currently, through the development of nano science, silica particles are being widely used as the most universal and basic material in such areas as bio sensors, bio imaging, contrast media for magnetic resonance imaging (MRI) devices, and bio medical areas of drug storage and carriers. This is due to silica’s role as a joining medium for dual coating and binding with metal materials including iron (Fe), gold (Au), silver (Ag), and nickel (Ni) and is based on silica’s high biocompatibility and chemical stability, and also due to the numerous silanol groups (Si-OH) that can form on surfaces,3-5) and that can be used with materials such as alumina (Al2O3) and ceria (CeO2) as a source material for Si-based photovoltaic modules and in the CMP (chemical mechanical polishing) process, in which flatly polished thin films formed unnecessarily on the wafer surface in the semiconductor process are produced through coating of special materials.6-8) Such silica particles, widely applied in the energy, environment, electronic and bio areas, have seen a gradual increase in the required values of characteristics for the particles themselves, in order to satisfy required characteristic values including characteristics for the control of the shape for uniform, spherical and mono-dispersed silica particles, as well as characteristics for the fine control of the size.9) Silica particles allowing sufficient control to form uniform spherical shape and size have advantages of allowing enhancement of the possibility of a role for these materials as a medium for joining with dual metals through high specific surface areas, and for improvement of efficiencies in the polishing process through formation of silica particles having uniform spherical shape and high purity. However, particle shapes resulting from gradual refinement of the particle size have been found to exhibit a phenomenon in which initial particles gradually aggregate upon synthesis through Van der Waals force or Gibbs free energy, causing difficulties in obtaining uniform particles.
Representatively, this problem occurs upon manufacturing of silica particles based on water glass, etc. as an inexpensive material. Even though synthesized through a simple synthesis process, such synthesized materials, which are of low efficiency, have been manufactured and have been found to have problems in which the yield of the outputs was low compared with the amount of incorporated raw materials and the particle shapes were non-uniform. In solutions for these problems, additional problems were encountered in which sufficient reaction times and processes under ancillary temperature conditions had to be incorporated to yield particles at a high efficiency; this was the case even though yield of uniform particles and control for finer sizes were possible through the solution measures of the manufacturing method, which is based on silica precursors for tetraethylorthosilicate (TEOS) and tetramthylorthosilicate (TMOS). Also, an additional cleaning process was necessary because the process required the use of inorganic solvents, which bring problems of non-eco-friendliness to the process of waste water processing.
In the present study, such problems are recognized, and particles with mono-dispersed spherical shape and high purity are to be developed through incorporation of simplified processes in a water soluble solvent, rather than in an organic solvent, as well as through the application of fast reaction times. In addition, the optimum process mechanism is to be presented, with a focus on the changes occurring in the growth of silica particles via the control of parameters for selective control of the size of the manufactured particles. Silica particles will be synthesized using the “Stöber method” of the sol-gel process, leading to many advantages for material synthesis due to the characteristics of the low-temperature process. Upon addition of TEOS as a precursor for the formation of silica particles and synthesis, sol is converted to gel through a continuous hydrolytic condensation reaction of Si-OH coupling by substitution of an alkoxide group (-OR) with a hydroxyl group (-OH). This is the result of the provision of a base environment and a change in the pH environment: spherical silica particles are formed layer by layer through continuous ionic coupling. Through control of the contents of distilled water and ammonia, and of the RPMs, the effects of the variables of the thus-formed particles on the particles will be observed as functions of the type and concentration change of the solvent upon synthesis, the concentration change of the catalyst used, and the stirring speed upon reaction.
2. Experimental Procedure
For spherical silica nano particles, 400-700 mL of distilled water (DIW, Milli-Q Pure water System, United States) was mixed for 15 minutes under each set of conditions after fixing 500 mL of ethanol (EtOH, Samchun Pur Chemical, Republic of Korea) in a 3-necked round flask as the reaction container. After incorporating 200 mL of tetraethylorthosilicate (TEOS, 95.0%+, Sigma Aldrich, United States) reagent as the precursor for the silica particles and mixing for 30 minutes, 30-50 mL of ammonia (NH4OH, Samchun Pure Chemical, Republic of Korea) solution was incorporated to form the base atmosphere required upon hydrolysis of reactants; this was followed by stirring at 450 - 650 RPM for the reaction time of 120 minutes. Conditions per composition are as shown in Table 1 and are also presented below. Silica particles prepared according to each composition were subjected to ultrasonic treatment for 10 minutes after incorporation of 500 mL of ethanol to remove the base reagent from the reactant and obtain the pure particles. The treated particles were then refined for 15 minutes in a centrifugal separator (Centrifuge, HANIL Science Industrial, Combi 514R, Republic of Korea) at 10,000 RPM. After cleaning by repeating the above process 3 times, the samples were frozen at −35°C by using a freeze dryer (LAB-CONCO, Kansas City, MO, United States); samples were then cooled to the cold trap temperature of −70°C and the hotplate temperature of −25°C; this was followed by drying of the samples with completion of the freezing process via the operation of a vacuum pump, producing a final yield of pure silica particles.
To identify the forming status of the silica particles, X-ray diffraction (XRD, Ultima IV, Rigaku, Japan) and Fourier transform infrared spectroscopy (FT-IR, Nicolet 5700, Thermo Electron, United States) were employed, while field emission scanning electron microscopy (FE-SEM, JSM-7610F, JEOL, Japan) was used to check for the particle shape. Also, to measure the particle size distribution and the dispersion characteristics in aqueous solution of the prepared particles, comparative analysis was performed for zeta potential and poly dispersity index (NanoZS, Malvern, United Kingdom).
3. Results and Discussion
Figure 1 shows XRD patterns, which were used to check the crystal structures of the prepared silica particles as the synthetic product. As a whole, peaks of wide area were observed in the region of 20 ~ 25 degrees, and the presence of no crystallographic planes was confirmed in the additional regions.10) Using the inherent identification number for SiO2 of the JCPDS (Joint Committee on Powder Diffraction Standards) Card (No.47-0715), the peak regions inherent to silica particles were confirmed, and particles having the form of an amorphous structure were considered to have been formed. To check the additional validity status of the silica particles, as in the XRD analysis, FTIR spectra were used for measurement. The synthesized samples were analyzed through the selection of the H7N5R45 synthesis sample, shown in Fig. 2, according to the fact that the X-ray diffraction analysis for the synthetic sample showed generally the same results for crystallographic planes. In the case of the H7N5R45 sample, Si-O-Si stretch was confirmed in the wave number region of 1,178 cm−1, with the wave number region of 948 cm−1 being confirmed for Si-OH stretch. In addition, the characteristics of Si-O-Si bending were observed at the 788 cm−1 peak for Si-OH and at the 3,411 cm−1 peak for the -OH group. This inherent region of wave number is the representative peak region of SiO2; pure silica particles are considered to have been formed because there was no observation of additional regions. In the case of the prepared particles, Si-O coupling is a quadruple coupling, which is considered to grow layer by layer and to form a structure retaining a lot of hydroxyl (-OH) groups at the end.11) Through observation of the amorphous structure and the region of wavelength number inherent to silica particles in the results shown in Fig. 1 and Fig. 2, the validity status of the silica particles was confirmed.
Figure 3 shows microstructure images used for analyzing the shape of the particles prepared as a function of the added ratio of distilled water. In general, the shape of particles that was observed was spherical, without aggregation; average particle sizes of (a) 280 ~ 300 nm, (b) 200 ~ 230 nm, (c) 150 ~ 180 nm and (d) 100 ~ 120 nm were measured. Differences in average size as a function of individual conditions suggest that the particle size could be controlled by controlling the amounts of distilled water added. When the amount of distilled water was reduced to 400 mL, the particle size showed an increase up to a maximum of 300 nm; a maximum size reduction of 100 nm was confirmed in the case of an amount of water added of 700 mL. Based on these observations, it is thought that small and uniform particles can be obtained through the effects where the rates of hydrolysis reaction with TEOS are increased; this will lead to an increase in the amount of H2O upon formation of an Si-O quadruple coupling structure. Also, at high amounts of water added, the generation of nuclei for the majority of silica particles can be more rapidly realized than is the case when the amounts of distilled water are smaller.12) However, no further reduction in particle size was observed even when the amount of distilled water was increased to more than 700 mL. This suggests that the amount of distilled water reacting with TEOS upon generation of particle nuclei is set so that distilled water of more than a given amount cannot be involved in the formation of particles.
Figure 4 shows FE-SEM images as a function of reduction in contents when ammonia is selected for the role as a catalyst capable of forming a salt atmosphere and of inducing a hydrolysis reaction. In Fig. 3, the particles generally showed uniform spherical shapes, and the reduction in average particle sizes from (a) 80 nm to (b) 50 nm was confirmed as a function of the reduction in the contents. Here, ammonia ions and TEOS in the aqueous solution become a state of weak acid and weak base, respectively, inducing a hydrolysis reaction, in which control of the hydrolysis/condensation reaction was confirmed to be possible through reduction in the ratio for ammonia contents. Through this, the particles maintain uniform shape, allowing size reduction even upon addition of the same amount of distilled water. However, as with the increase in the amount of distilled water, no change in particle size was observed upon further reduction of the content of the ammonia solution to less than 30 mL, and particle formation was not realized upon additional reduction. This confirms the fact that ammonia to promote the condensation reaction is the essential element for not only particle size but also formation of uniform particles. Also, in the case of particle (c), non-uniform shapes and sizes of particles were confirmed as aggregation occurred up to the size of 200 nm ~ 1 μm, when the added amount of ammonia was increased to 60 mL, while aggregation became severe upon increase of (d) 70 mL, leading to a particle size larger than 1 μm. These results suggest that the process of sol-to-gel conversion is promoted for aggregation as the condensation reaction accelerates more than the hydrolysis reaction, and when the ammonia content is increased to more than a given amount during the synthesis reaction.13) Consequently, as with control of distilled water, control of ammonia is considered to be the essential element for further refinement of the hydrolysis and condensation reaction on the basis of the same time.
Figure 5 provides images representing the graphs of the average particle diameter of the silica particles prepared as a function of changes in distilled water and ammonia. In the graph, curves with mono-modal tendency for each condition were confirmed; as a result of control through distilled water, the measured average sizes are shown to be (a) 275 nm, (b) 213 nm, (c) 186 nm and (d) 114 nm, as can be seen in Fig. 3. Also, the average particle size distributions of (a) 82 nm and (b) 51 nm are recorded in Fig. 4; these samples were prepared as a function of the ammonia variables. These graphs are considered to show the same average particle size as the uniform particle size, as do the silica particles dispersed in aqueous solution, which impede aggregation among particles through the presence of a lot of surface hydroxyl (-OH) groups, as can be seen in Fig. 2.14) In addition, the graph can be confirmed to show broader curves when the average particle diameter of the silica particles is larger. Based on these observations, this form of the graph considered to occur because the suspension in use for this measurement is H2O, such that the SiO2 particles contain H2O.
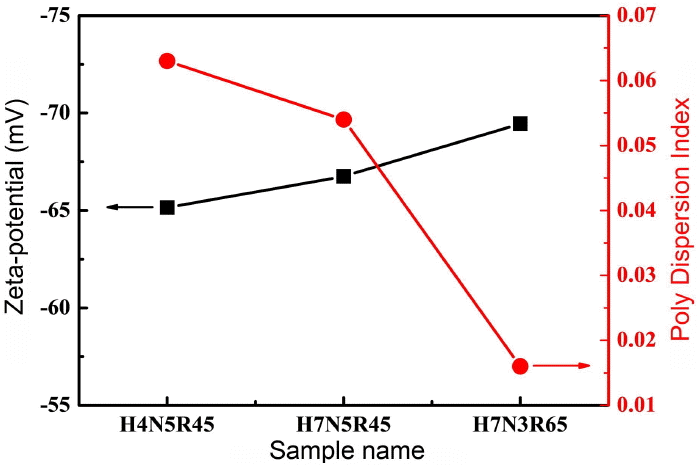
Figure 6(a) provides an SEM image for the sample prepared through control at 650 RPM. For the initial particles synthesized under the condition of 650 RPM, a mesh-type structure appearing to show severe aggregation among particles can be confirmed by visual check. This structure appears to have been induced by the high surface energy of the initial particles, which was the result of a reduction in the particle size. However, the average size of the particles was shown to be 32 nm when the average particle diameter in the aqueous solution of (b) was confirmed. To digitize and analyze the surface charge of the particle surfaces through these results, results of zeta potential and poly dispersity index measurements, which are shown in Fig. 7, were obtained. Each sample was measured under the same condition in the water soluble suspension (pH 6.8 ~ 7.0); the samples controlled to have sizes of 275 nm, 114 nm and 30 nm were selected. For the zeta potentials of each sample, high negative (−) figures of surface charge at (a) −65.15 mV, (b) −66.75 mV and (c) −69.45 mV were recorded. This can provide a supplementary description for the measured results shown in Fig. 6. As a result, although the small initial particles are to be mutually aggregated to exist in the form of large clusters for stabilization due to the action of the Van der Waals force among the particles, the prepared silica particles have an electrostatic repulsive force due to the high negative charge, allowing a stable distance to be maintained without aggregation. Also, the mono-disperse index (DI) values for each specimen were confirmed to be smaller than 0.1 at 0.063, 0.054 and 0.016, and the silica particles of the sample formed according to the reduction width of the particles are considered to be mono-dispersed in the aqueous solution.15) Hence, the average sizes of the particles can be controlled up to the maximum of 32 nm.
4. Conclusions
Presented in the present study is a development method for silica particles simplified within a hydrophilic solvent through the Stöber method of the sol-gel process. For silica particles prepared through application of fast reaction time and incorporation of simplified processes, different variables of the content ratios for distilled water and ammonia, and different values of RPM were applied. For these particles, as a function of the content ratio of distilled water, size control in a range of 100 ~ 300 nm was possible, while sizes of 50 ~ 80 nm were observed for particles prepared as a function of the content ratio of ammonia. Also, formation of the particle size could be controlled down to a minimum of 32 nm through an increase in the reaction RPMs. To check for cohesiveness of the particles prepared at sizes down to 32 nm, zeta potential and poly dispersity index analyses were performed by re-dispersion of the dried silica particles in the suspension state. First, excellent dispersion figures of −60 ~ −70 mV for surface charge, and measured values of 0.063 ~ 0.016 of high poly dispersity index, which are under a value of 0.1, were confirmed. As a result, although with a reduction in the particle size there is a tendency toward stabilization by aggregation in the cluster form due to the Van der Waals force, the electrostatic repulsive force acts among particles through retention of a lot of hydroxyl (−OH) groups at the surface ends in the prepared silica particles. This impedes aggregation among particles, allowing stabilized particles to be obtained. Based on the present study, mono-dispersed spherical silica, allowing selective size control in a range from 30 ~ 300 nm, is considered to be suitable for industry-wide use.