The Effect of Domain Wall on Defect Energetics in Ferroelectric LiNbO3 from Density Functional Theory Calculations
Article information
Abstract
The energetics of defects in the presence of domain walls in LiNbO3 are characterized using density-functional theory calculations. Domain walls show stronger interactions with antisite defects than with interstitial defects or vacancies. As a result, antisite defects act as a strong pinning center for the domain wall in LiNbO3. Analysis of migration behavior of the antisite defects across the domain wall shows that the migration barrier of the antisite defects is significantly high, such that the migration of antisite defects across the domain wall is energetically not preferable. However, further study on excess electrons shows that the migration barrier of antisite defects can be lowered by changing the charge states of the antisite defects. So, excess electrons can enhance the migration of antisite defects and thus facilitate domain wall movement by weakening the pinning effect.
1. Introduction
Ferroelectric materials are of interest for such diverse applications as non-volatile Ferroelectric Random Access Memory (FRAM),1) electro-optic modulators2) and frequency converters.3) Especially, LiNbO3 has excellent piezoand pyro-electricity properties, is photo-refractive, and displays nonlinear optical properties. It also has a high spontaneous polarization, 70 μC/cm2, and high Curie temperature, ~1480 K,4,5) so it is suitable for high temperature applications as well. Generally, congruent LiNbO3, which has an R3c structure, is easy to grow under Li deficient conditions. 6,7) However, recent successes using the vapor-transport equilibration (VTE) method8) have enabled the growth of stoichiometric LiNbO3. Although various defect models including those for pseudo-Schottky defects, Schottky defects, and Frenkel defects have been suggested9) to explain energetically preferred defects and defect clusters in congruent LiNbO3, only an Nb antisite, compensated for by four Li ion vacancies9–11) and five Nb antisites compensated for by four Nb vacancies11) were found to successfully explain the stoichiometry of congruent LiNbO3. Atomistic modeling by Donnerberg et al.12) showed that compensation by Nb vacancies is energetically less favorable than compensation by Li vacancies. Recent DFT calculations13) showed that Nb antisite compensated for by Li vacancies, and the Li Frenkel pair, are the most energetically favorable defect clusters in LiNbO3 under Li deficient (congruent) and Li rich (stoichiometric) conditions, respectively. An analysis on various Nb antisite-Li vacancy cluster models determined that several arrangements of Li vacancies around an Nb antisite are nearly energetically equivalent.14)
Interfaces between two opposite polarization domains (Domain walls) are also studied using both experiments and simulations. Gopalan et al.15) suggested two different domain walls, the Y- and X-walls, based on crystallographic orientation; the Y-wall lies parallel to the (112̄0) plane, while the X-wall lies parallel to the (101̄0) plane. In subsequent works,16) the X-wall was identified as a mixed anion-cation plane, while the Y-wall was determined to be alternating planes of cations only and anions only. Energetic studies on the two domain walls17) show that the Y-wall is energetically favored but less mobile. It was also found that non-uniaxial polarization components near the domain walls can lead to Bloch-like characteristics of the X-wall and both Bloch-like and Néel-like characteristics of the Y-walls. 18) Previous studies have demonstrated that defect/domain wall interactions can lead to switching in the preferred orientation of the domain wall from the Y-wall to the X-wall19) or to a reduction of the electric polarization.20) In extending the previous work on defects and domain walls, this study mainly focuses on the interaction between defects and domain walls in LiNbO3 and the effect of those defects on the domain dynamics. Various aspects of defects, including type and charge state, are investigated to understand the effect of domain walls on defect energetics.
2. Simulation Methods
All the calculations are performed within the density functional theory (DFT)21) level with generalized gradient approximation22) of exchange and correlation potential. Monkhorst-Pack23) k-point sampling with a grid of 1 × 4 × 2 is used. The plane wave DFT calculations are performed with the Vienna Ab Initio Simulation Package (VASP)24,25) using the projected augmented wave (PAW)26) pseudopotential. The generalized gradient approximation (GGA)22) is used to evaluate the exchange and correlation interactions. The plane waves are expanded up to 400 eV of kinetic energy.27) Ionic relaxation is performed until the maximum atomic force is below 0.01 eV/Å. The self-consistent solution of the Kohn-Sham functional is obtained using the residual minimization method direct inversion in the iterative subspace (RMM-DIIS) algorithm,28) which optimizes several individual energy bands at the same time. The pseudopotential and methodologies used here are the same as those used in our previous studies of intrinsic defects13) and domain walls.17) The climbing-image Nudged Elastic Band (NEB) method29,30) with 10 images is employed to predict the energy barrier of defect migration.
3. Results and Discussion
Although the ferroelectric fatigue and optical instability of ferroelectric LiNbO3 are known to result from defects/domain wall interactions, these drawbacks still limit the usage of ferroelectric materials. To overcome the current application limits, thorough understanding of the interaction mechanism is necessary. In the current study, the interaction energetics are determined by putting three major point defects
Figure 1 provides a schematic view of the Y-wall in LiNbO 3 and the spatial location of each point defect near the Y-wall. As can be seen, the Y-wall sits between two atom planes; point defects are located at different distances from the domain walls. For all cases, the defect/domain wall interactions decreased the energy, indicating that defects are preferentially positioned near the domain wall. For the Y-wall, the quadrupley charged
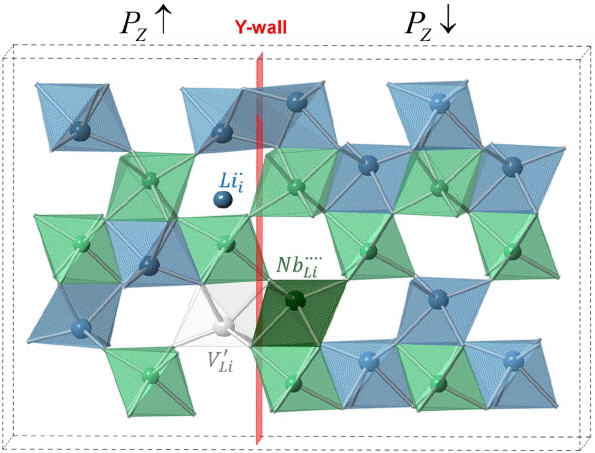
Schematic view of Y-wall and three major points defects in LiNbO3;
A similar study has been also performed for the X-walls. Because our previous study identified two inequivalent X-walls related with the oxygen sublattice, current studies have calculated the interaction energies of intrinsic defects with both the XI-wall and the XII-wall. Although maximum interactions are observed within the second nearest plane from both the X-walls regardless of the charge states of the intrinsic defects, the energetics for their interactions are different. Fig. 2 shows a comparison of the maximum defects/X-wall interaction energy within the three nearest planes. As can be seen in Fig. 2, the majority of defects show stronger interaction with the XI-wall than with the XII-wall. The most prominent difference in the interaction energetics is observed for the quadrupley charged
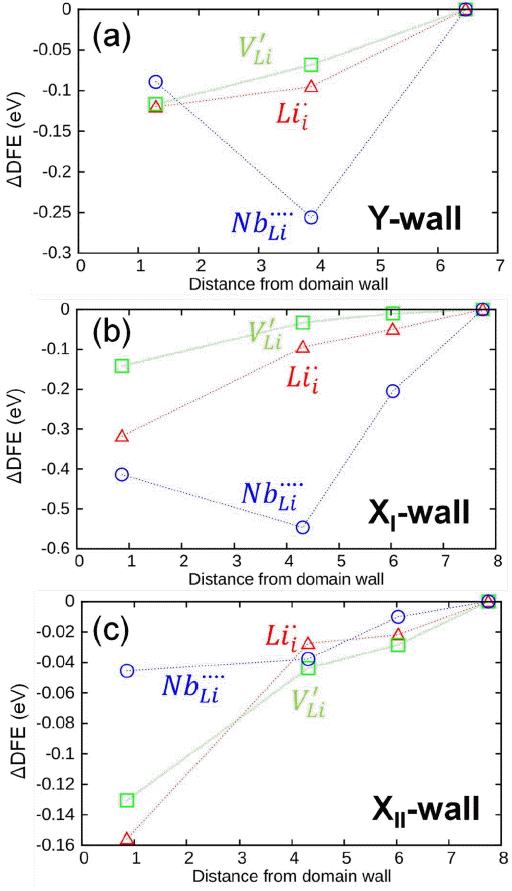
Variation of defect formation energy (DFE) as a function of distance from (a) Y-wall, (b) XI-wall and (c) XII-wall. The DFE of three dominant point defects:
Previous study has shown that the domain wall in PbTiO332) acts as a trap site for oxygen vacancies. As an extension of that study, the current work shows that the domain wall does not act as a trap site for only oxygen vacancies, but for all other intrinsic defects regardless of their charge states or defect types. For the Y-wall, the quadrupely charged
Because the domain walls act as trap sites for defects, defects might form a cluster near the domain walls. In order to verify the energetics of single defect/domain wall interactions, we have also considered defect clusters. For this study, we have considered two different defect pairs,

Comparison of Interaction Energies of Two Defect Complexes with One Y-wall and two X-walls. The DFE at Bulk State is Shown as a Reference for Each Configuration
We have looked at the energetics of the defects interacting with domain walls in LiNbO3. Now we will discuss how the defects can migrate near the domain wall. In order to study the migration of various point defects, we have employed the NEB method along the migration pathway. Because the previous study17) has shown that the Y-wall, lying parallel to the (112̄0) plane, is energetically much more favorable than the two X-walls, our study focused on the migration behavior of defects around the Y-wall. Especially, the current study has focused on the

Potential energy profile along the migration pathway of the
We now consider how the migration behavior of the

Potential energy profile along the migration pathway of
The addition of an extra electron decreases the migration barrier of the
4. Conclusions
Current works have performed a systematic study of the energetics of defects/domain wall interaction in LiNbO3. Our energetic study predicts that the formation energy of intrinsic defects can be lowered near the domain wall. So, the interactions of intrinsic point defects with domain walls decrease the DFE and trap the defects in the domain wall. In the following study, we find that the migration barrier of
Acknowledgments
This work was supported by Chonnam National University, 2015, and by the National Institute of Supercomputing and the Network/Korea Institute of Science and Technology Information with supercomputing resources including technical support, No. KSC-2015-C3-034.